Drug Dose Response Curves: Fitting and Plotting
Jermiah J. Joseph
Princess Margaret Cancer Centrejermiah.joseph@uhn.ca
18 October 2024
Source:vignettes/CurveFitting.Rmd
CurveFitting.RmdGenerate Profiles for a Single Sample and Treatment
pset <- CBWWorkshop2024::dummy_pset
show(pset)
#> <PharmacoSet>
#> Name: dummy_pset
#> Date Created: Tue Oct 15 23:31:41 2024
#> Number of samples: 50
#> Molecular profiles: <MultiAssayExperiment>
#> ExperimentList class object of length 2:
#> [1] rnaseq.tpm : SummarizedExperiment with 100 rows and 50 columns
#> [2] rnaseq.tpm.batch : SummarizedExperiment with 100 rows and 50 columns
#> Treatment response: <TreatmentResponseExperiment>
#> dim: 4847 52
#> assays(1): sensitivity
#> rownames(4847): 1e-07:Ixabepilone:1 1e-06:Ixabepilone:1 ... 1000:Nelarabine:1 4000:Ifosfamide:1
#> rowData(4): treatmentdose treatmentid tech_rep CONC1
#> colnames(52): 786-O:1 A-498:1 A-549:1 ... UACC-257:1 UACC-62:1 UO-31:1
#> colData(3): sampleid bio_rep PANEL
#> metadata(0): none
SAMPLE_OF_INTEREST <- "786-O"
TREATMENT_OF_INTEREST <- "Ixabepilone"
pset |>
# Get the TreatmentResponseExperiment
treatmentResponse() |>
# Filter the data to a single sample and treatment
subset(
treatmentid %in% TREATMENT_OF_INTEREST,
sampleid %in% SAMPLE_OF_INTEREST
) |>
# Group By to average out technical replicates
CoreGx::endoaggregate(
viability = mean(viability),
assay = "sensitivity",
target = "mean_sensitivity",
by = c("treatmentid", "treatmentdose", "sampleid", "bio_rep")
) |>
# Fit the dose-response curve
CoreGx::endoaggregate(
{
fit <- PharmacoGx::logLogisticRegression(treatmentdose, viability)
IC50 <- PharmacoGx::computeIC50(treatmentdose, Hill_fit = fit)
AAC <- PharmacoGx::computeAUC(treatmentdose, Hill_fit = fit, area.type = "Fitted")
list(
HS = fit[["HS"]], E_inf = fit[["E_inf"]] / 100, EC50 = fit[["EC50"]],
Rsq = as.numeric(unlist(attributes(fit))),
AAC = AAC,
IC50 = IC50,
min_dose = min(treatmentdose),
max_dose = max(treatmentdose),
max_val = max(viability),
min_val = min(viability)
)
},
assay = "mean_sensitivity",
by = c("treatmentid", "sampleid", "bio_rep"),
enlist = FALSE,
target = "profiles"
) -> tre_profiled
tre_profiled
#> <TreatmentResponseExperiment>
#> dim: 35 1
#> assays(3): sensitivity mean_sensitivity profiles
#> rownames(35): 1e-07:Ixabepilone:1 1e-06:Ixabepilone:1 ... 0.3:Ixabepilone:7 0.3:Ixabepilone:8
#> rowData(4): treatmentdose treatmentid tech_rep CONC1
#> colnames(1): 786-O:1
#> colData(3): sampleid bio_rep PANEL
#> metadata(0): noneHelper function to plot the curve
#' @title Plot a dose-response curve
#'
#' @description This function plots a dose-response curve for a given sample and treatment
#'
#' @details This function takes a `CoreGx::TreatmentResponseExperiment` object and plots a dose-response curve for a given sample and treatment. The curve is fitted using the Hill equation.
#'
#' @param tre `CoreGx::TreatmentResponseExperiment` object
#' @param sampleid `character` sample id
#' @param treatmentid `character` treatment id
#' @param raw_assay_name `character` name of the raw assay
#' @param profiles_assay_name `character` name of the profiles assay
#' @param HS_colname `character` name of the HS column
#' @param E_inf_colname `character` name of the E_inf column
#' @param EC50_colname `character` name of the EC50 column
#'
#' @return NULL
#'
#' @export
#'
plot_curve <- function(
tre,
sampleid,
treatmentid,
mono_assay_name = "sensitivity",
profiles_assay_name = "profiles",
HS_colname = "HS",
E_inf_colname = "E_inf",
EC50_colname = "EC50"
){
mono_assay <- tre[[mono_assay_name]][
treatmentid == treatmentid & sampleid == sampleid
]
profile_assay <- tre[[profiles_assay_name]][
treatmentid == treatmentid & sampleid == sampleid
]
min_dose <- profile_assay$min_dose
max_dose <- profile_assay$max_dose
pseudo_x <- seq(log10(min_dose), log10(max_dose), length.out = 100)
pseudo_y <- PharmacoGx:::.Hill(
pseudo_x,
c(
profile_assay[[HS_colname]],
profile_assay[[E_inf_colname]],
log10(profile_assay[[EC50_colname]])
)
)
df <- data.frame(
treatmentdose = log10(mono_assay$treatmentdose),
viability = mono_assay$viability / 100
)
pseudo_df <- data.frame(
pseudo_x = pseudo_x,
pseudo_y = pseudo_y
)
# plot
library(ggplot2)
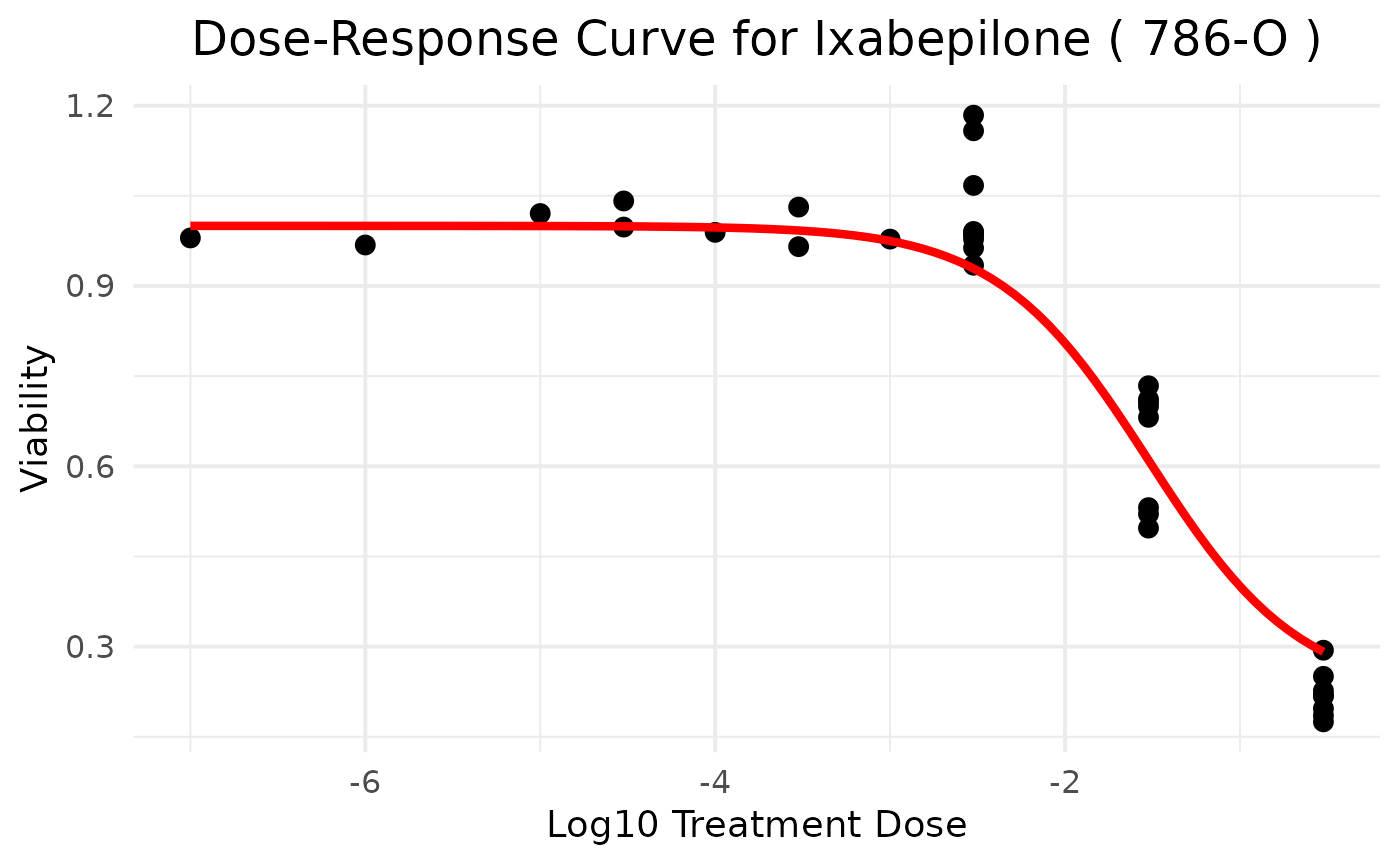
ggplot() +
geom_point(data = df, aes(x = treatmentdose, y = viability), color = "black", size = 3) + # Points
geom_line(data = pseudo_df, aes(x = pseudo_x, y = pseudo_y), color = "red", size = 1.5) + # Fitted curve
labs(
title = paste("Dose-Response Curve for", treatmentid, "(", sampleid, ")"),
x = "Log10 Treatment Dose",
y = "Viability"
) +
theme_minimal(base_size = 15) +
theme(
plot.title = element_text(hjust = 0.5),
axis.text = element_text(size = 12),
axis.title = element_text(size = 14)
)
}